Bot. Bull. Acad. Sin. (1998) 39: 23_27
Rout et al. Genetic stability of micropropagated plants of ginger
Determination of genetic stability of micropropagated plants of ginger using Random Amplified Polymorphic DNA (RAPD) markers
G.R. Rout1,3, P. Das1, S. Goel2, and S.N. Raina2
1Plant Biotechnology Division, Plant Tissue Culture Laboratory, Regional Plant Resource Centre, Bhubaneswar - 751 015, Orissa, India
2Plant Cytogenetics and Molecular Biology Laboratory, Department of Botany, Delhi University, Delhi - 110 007, India
(Received July 16, 1997; Accepted October 3, 1997)
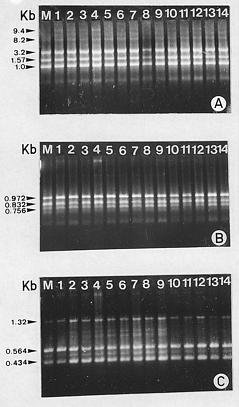
Abstract. Random amplified polymorphic DNA (RAPD) markers were used to evaluate the genetic stability of micropropagated plants of Zingiber officinales cv. V3S18. Fifteen arbitrary decamers were used to amplify DNA from in vivo and in vitro plant material to assess the genetic fidelity. All RAPD profiles from micropropagated plants were monomorphic and similar to those of field grown control plants. No variation was detected within the micropropagated plants. The utilization of RAPD markers both for the assessment of genetic stability of clonal materials and to certify genetic stability throughout the systems of micropropagation is discussed.
Keywords: Genetic markers; Genetic stability; Micropropagation; RAPD.
Introduction
Zingiber officinales Rosc. (ginger) of the family Zingiberaceae is an important tropical horticultural plant, valued all over the world as a spice in culinary preparations and for its medicinal properties. It is rich in secondary metabolite such as Oleoresin (Bhagyalakshmi and Singh, 1988). Breeding of ginger is seriously handicapped by poor flowering and seed set. Most crop improvement programs of this species are confined to evaluation and selection of naturally occurring clonal variations. In vitro culture techniques provide an alternative means of plant propagation and a tool for crop improvement (Vasil, 1988). Clonal multiplication of ginger through shoot multiplication has been reported (Hosoki and Sagawa, 1977; Wang, 1989; Balachandran et al., 1990; Rout et al., 1997). Plantlets derived from in vitro culture might exhibit somaclonal variation (Larkin and Scowcroft, 1981) which is often heritable (Breiman et al., 1987). Other reports claim that useful morphological, cytological, and molecular variations may be generated in vitro (Larkin et al., 1989). Any system which significantly reduces or eliminates tissue culture generated variations can be of much practical utility. The variations may be due to several factors (Vasil, 1987; 1988), such as genotypes used (Breiman et al., 1987), pathways of regeneration, and parameters employed for assessing the effect of in vitro culture, such as gross morphology and cytology (Swedlund and Vasil, 1985), field assessment, and molecular studies (Breiman
et al., 1989; Chawdhury et al., 1994; Shenoy and Vasil, 1992).
Several strategies can be used to assess the genetic fidelity of in vitro derived clones, but most have limitations. Karyological analysis, for example, cannot reveal alterations in specific genes or small chromosomal rearrangements (Isabel et al., 1993). Isozyme markers provide a convenient method for detecting genetic changes, but are subject to ontogenic variations. They are also limited in number, and only DNA regions coding for soluble proteins can be sampled. Using the polymerase chain reaction (PCR) in conjuction with short primers of arbitrary sequence (Williams et al., 1990), randomly amplified polymorphic DNA (RAPD) markers were recently shown to be sensitive for detecting variations among individuals between and within species (Carlson et al., 1991; Roy et al., 1992). RAPD markers have been used successfully to assess genetic stability among somatic embryos in spruce species (Isabel et al., 1993; 1996) and among micropropagated plants of poplar (Rani et al., 1995). The present study was undertaken to determine the genetic stability of the micropropagated plants of Zingiber officinales cv. V3S18 by RAPD analysis.
Materials and Methods
The standard protocol of micropropagating Zingiber officinales cv. V3S18 plants through meristem culture was developed and tested at the Regional Plant Resource Centre, Bhubaneswar (Rout et al., unpublished). The protocol was used for assessment of genetic stability. In the present
3Corresponding author. Fax: 91-0674-450274.