Lin et al. Differentially expressed cDNAs from mungbean
Bot. Bull. Acad. Sin. (1998) 39: 87_91
Rapid isolation of differentially expressed cDNAs from near isogenic lines of mungbean
Ching-Yu Lin1, Cheng-Chun Kuan1, Rong-Huay Juang1, Samson C. S. Tsou2, C. George Kuo2 and Ching-San Chen1,3,4
1Graduate Institute of Agricultural Chemistry, National Taiwan University, Taipei, Taiwan, Republic of China
2Asian Vegetable Research and Development Center, P.O. Box 42, Shanhua, Tainan 741, Taiwan, Republic of China
3Institute of Botany, Academia Sinica, Nankang, Taipei, Taiwan, Republic of China
(Received August 4, 1997; Accepted November 28, 1997)
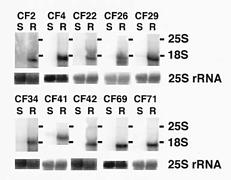
Abstract. We describe an application of cDNA libraries constructed from total RNA to isolate simultaneously many differentially expressed cDNAs between a pair of near isogenic lines of mungbean in a short period of time. All ten selected individual cDNA clones, containing inserts ranging from 1.1_1.9 kb, clearly showed positive Northern blotting. Nucleotide sequencing indicated that six of the clones were probably full-length cDNAs.
Keywords: Differentially expressed cDNAs; Mungbean; Near isogenic line.
Introduction
RNA fingerprint by arbitrarily primed PCR (Welsh et al., 1992) and differential display of RNA (Liang and Pardee, 1992) have been successfully used to isolate differentially expressed genes in two different states of many biological systems. Both techniques are powerful and require only small amounts of total RNA, but they also share some limitations. First, these methods produce a rather high rate of false positives, in which usually more than one cDNA species is present within one amplified band. Therefore, cloning a gene from the cDNA mixture is difficult. Consequently, obtaining cDNA clones that show positive results in Northern blotting is laborous and time-consuming. Various strategies have been tried to circumvent this problem (Mathieu-Daude et al., 1996; Zhao et al., 1996; Zhang et al., 1996; Shoham et al., 1996; Poirier et al., 1997). Secondly, it is impossible to obtain a full length cDNA by these methods, because of the limitation of the DNA sequencing gel that allows determination of only a maximal length of 500 bp. A full length cDNA has to be isolated from a cDNA library or by means of a RNA-anchored cDNA extension (RACE) method. Thirdly, these methods use radioisotopes. In this paper, we describe a rapid method based on the CapFinder Kit to simultaneously isolate many differentially expressed cDNAs without the use of radioisotopes.
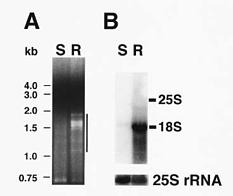
In our study on the isolation of differentially expressed full-length cDNAs from a pair of near isogenic lines of mungbean, it was found that cDNA bandings generated by the CapFinder Kit were directly visualized on an agarose gel. No radioactivity was required to estimate yield or
size distribution. It was also found that the differentially expressed cDNAs from the near isogenic lines were resolved nicely on the agarose gel. As a result, some differentially expressed cDNA species were recovered from the agarose gel and subcloned. The individual cloned cDNA showed positive results in Northern blot analysis, and their nucleotide sequences were determined. This method seems particularly useful in the isolation of differentially expressed abundant genes in a short period of time.
Materials and Methods
Plant Varieties
Vigna radiata L. VC 1973A and VC 6089A were bred in the Asian Vegetable Research and Development Center (AVRDC). VC 6089A (BC6S2) populations were crossed using a wild mungbean TC 1966 as donor parents and breeding lines VC 1973A as recurrent parents.
RNA Preparation
Total RNA was extracted from the 15 DAF developing seeds of VC 1973A and VC 6089A by hot phenol method (Verwocrd et al., 1989). DNA-free total RNA was prepared as described previously (Oh et al., 1995).
Preparation of Full-Length Double-Stranded cDNA
The total RNAs were used to synthesize first and second-strand cDNAs using a CapFinderTM PCR cDNA library construction kit (Clontech). For the first step of the procedure, a 10 ml reverse transcription reaction mixture contained 1 × first-strand buffer, 10 mg of DNA-free total RNA, 1 mM of CapSwitch oligonucleotide (5'-TGCTGCGGAAGACGACAGAAGGG-3'), 1 mM of
4Corresponding author. E-mail: bocschen@ccvax.sinica.edu.tw