Bot. Bull. Acad. Sin. (2000) 41: 113-120
Shih et al. DNA probe for Xanthomonas campestris
A DNA probe for identification of Xanthomonas campestris pv. campestris, the causal organism of black rot of crucifers in Taiwan
Hsin-Der Shih1, Yuan-Chuen Lin1, Hsiou-Chen Huang2, Kuo-Ching Tzeng1, and Shih-Tien Hsu1,3
1Department of Plant Pathology, National Chung Hsing University, Taichung, Taiwan, Republic of China
2Graduate Institute of Agricultural Biotechnology, National Chung Hsing University, Taichung, Taiwan, Republic of China
(Received April 7, 1998; Accepted September 7, 1999)
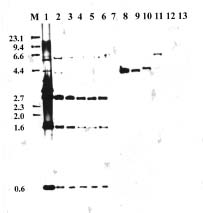
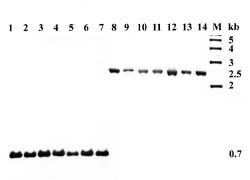
Abstract. A DNA probe was developed for identification of strains of Xanthomonas campestris pv. campestris from Taiwan. EcoRI restriction fragments of total DNA from X. campestris pv. campestris strain Xcc70 were cloned into pBluescript II KS and transformed into Escherichia coli DH10B. The recombinant plasmid DNAs from clones randomly selected were labeled with digoxigenin and screened for their specificity for X. campestris pv. campestris. In Southern hybridization, one of the clones (pXcc70-8) hybridized to several EcoRI-digested fragments of total DNA from only strains of X. campestris pv. campestris. Digestion of the insert DNA of the clone pXcc70-8 with EcoRI yielded fragments of 2.7, 1.6 and 0.6 kb. When a subclone containing the 0.6 kb fragment was used as a probe (Xcc70-8-l), it hybridized with all 51 strains of X. campestris pv. campestris and all seven strains of X. campestris pv.armoraciae, but not with the 60 strains of other bacteria tested. This probe, however, distinguishably detected a single fragment of 0.7 kb in strains of X. campestris pv. campestris and a single fragment of 2.5 kb in strains of X. campestris pv. armoraciae when their total DNAs were digested with KpnI. The detection limits of the probe Xcc70-8-l for the amount of DNA was 25 pg and for the number of cells was about 6 × 104 CFU in dot blot assays. The colony and dot blot hybridizations with the probe were used to detect X. campestris pv.campestris in extracts of infected leaves of cabbage and seeds of several crucifers. The results indicate that the DNA probe can be used to detect X. campestris pv. campestris in plant tissues but probably not in seeds. The probe could be a useful tool for rapid identification of the pathogen in epidemiological studies in Taiwan.
Keywords: Crucifers (Brassica spp.); Detection; DNA probe; Identification; Xanthomonas campestris pv. campestris.
Introduction
Black rot of crucifers caused by Xanthomonas campestris pv. campestris (Pammel) Dowson is a destructive disease of worldwide importance (Williams, 1980). In Taiwan, black rot affects many cruciferous crops and can be observed at all growing seasons, particularly in most cabbage and cauliflower fields. The black rot pathogen is seed-borne (Walker and Tisdale, 1920; Williams, 1980; Schaad, 1982). Seed testing to ensure absence of the pathogen and seed treatments to eradicate the pathogen are important measures for preventing the occurrence of the disease (Williams, 1980). The success of these measures depends on an efficient and reliable method to detect the pathogen in seeds and to verify the efficacy of seed treatments. The pathogen also survives in infected crop debris, infested cruciferous weeds, and infected plant residues in soil (Alvarez and Cho, 1978; Schaad and White, 1974b; Schaad and Dianese, 1981). The importance of these sources of inoculum in the annual recurrence of
the disease in Taiwan remains to be investigated. This kind of ecological study also requires an effective method to detect the pathogen in fields.
Selective media and serology are the commonly used methods for the detection of X. campestris pv. campestris in seed, plant, or soil samples (Chun and Alvarez, 1983; Franken, 1992; Fukui et al., 1994; Schaad and White, 1974a; Schaad and Donaldson, 1980). The detection efficiency of selective media varies greatly with the source of samples. Most of the available selective media are not efficient for detection of the pathogen in seeds and soils under Taiwan conditions (Huang and Hsu, 1987). Furthermore, the identity of the suspect colonies on the media needs to be confirmed either by the pathogenicity test, which is time-consuming, or by serology. Serological assays are useful in the confirmation test for the isolated colonies (Schaad, 1979) and for direct detection of the pathogen in seeds (Franken, 1992; Schaad and Donaldson, 1980), infected leaves (Alvarez and Lou, 1985), and soil (Domen and Alvarez, 1978). Serology with monoclonal antibodies is particularly specific for the identification or differentiation of strains of X. campestris pv. campestris (Alvarez et al., 1985) and has been used suc
3Corresponding author. Fax: 886-4-2877585; E-mail: sthsu@mail.nchu.edu.tw