Bot. Bull. Acad. Sin. (2000) 41: 121-128
Lee and Wang Detection of R. solanacearum by PCR
The design of specific primers for the detection of Ralstonia solanacearum in soil samples by polymerase chain reaction
Yung-An Lee1 and Chi-Chung Wang
Department of Biology, Fu Jen Catholic University, Hsin Chuang 24205, Taipei, Taiwan, Republic of China
(Received March 24, 1999; Accepted October 1, 1999)
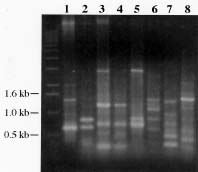
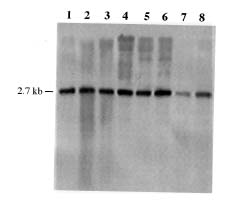
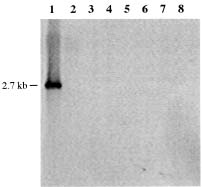
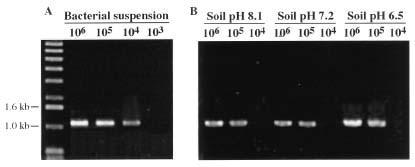
Abstract. A 0.7-kb DNA fragment, amplified by the randomly amplified polymorphic DNA (RAPD) method from Ralstonia solanacearum total DNA, was cloned and evaluated as a specific DNA probe. This 0.7-kb DNA fragment hybridized to a 2.7-kb EcoRI fragment in the EcoRI-digested total DNA of R. solanacearum. The 2.7-kb EcoRI fragment was also cloned and hybridized only to R. solanacearum but not to other Pseudomonas spp., pathovars of Xanthomonas campestris, and Erwinia spp. tested. The DNA sequence of this 2.7-kb fragment was obtained and used to design specific oligonucleotide primers for polymerase chain reaction (PCR) amplification. The primers amplified the same 1.1-kb PCR product from all R. solanacearum strains tested and failed to amplify DNA from any other plant pathogenic bacterial strains tested. DNA isolated from several saprophytic bacteria did not produce any PCR products with these primers. This specific PCR for R. solanacearum was also performed from colonies grown on BG medium with similar results. The sensitivity of the PCR assay using the specific primers was about 20 cells. The PCR assay was used to detect R. solanacearum in soil using these primer sets. No PCR product could be found when soil extract containing R. solanacearum was used directly in the assay. DNA extraction from soil was needed for the success of PCR assay. A simple method for DNA extraction from soil for PCR assay was developed and can hasten the detection of R. solanacearum in soil.
Keywords: Erwinia; Polymerase chain reaction; Pseudomonas; Ralstonia solanacearum; Xanthomonas.
Introduction
Ralstonia solanacearum (synonym Pseudomonas solanacearum E. F. Smith) (Yabuuchi et al., 1995) causes bacterial wilt, one of the most important and widespread bacterial diseases of crops in the world. This pathogen has been recorded on several hundred species representing 44 families of plants (Buddenhagen and Kelman, 1964; Hayward, 1991). In Taiwan, bacterial wilt has been reported on tomato, tobacco, potato, sweet pepper, eggplant, bird-of-paradise (Yang et al., 1980), perilla (Perilla crispa) (Hong et al., 1990), radish (Lin et al., 1994), anthurium plants (Su and Leu, 1995), and eustoma (Chao et al., 1995). Ralstonia solanacearum can be spread within and between countries by soil, by water, and by latently infected planting materials (Ciampi et al., 1980; Hayward, 1991). Accordingly, rapid and highly sensitive detection methods of R. solanacearum are required for quarantine to reduce field losses and limit the spread of bacterial wilt (Seal et al., 1992a, 1993; Seal, 1995).
The success of quarantine procedures relies on the use of rapid and sensitive detection techniques. Traditional methods for detecting R. solanacearum depend on a series of biochemical tests on purified colonies. This iden
tification procedure is too time consuming for quarantine and other diagnostic laboratory purposes. Nucleic acid probes and polymerase chain reaction (PCR), on the other hand, have been widely reported as tests that can be performed without culturing. Diagnostic tests based on PCR amplification of plant pathogens from environmental samples have shown that rapid and specific detection of very low numbers of the pathogen can be achieved (Seal et al., 1992a, 1993; Seal, 1995).
Development of a DNA probe specific for R. solanacearum is complicated by the genetic diversity of this species. Ralstonia solanacearum strains represent a heterogeneous group that has been subdivided informally into five races on the basis of host range (Buddenhagen et al., 1962; Buddenhagen, 1986), or five biovars based on the ability to oxidize certain disaccharides and hexose alcohols (Hayward, 1964; He et al., 1983). In addition, there is a phylogenetic dichotomy in R. solanacearum, which can be separated into two divisions based on the analysis of restriction fragment length polymorphism patterns (Cook et al., 1989), PCR amplification with tRNA consensus primers (Seal et al., 1992b), and rRNA sequences (Li et al., 1993; Seal et al., 1993; Seal et al., 1999; Taghavi et al., 1996). Division I contains all members of race 1 biovars 3, 4 and 5, and division II contains all members of race 1 biovar 1 and races 2 and 3. Strains of R. solanacearum in Taiwan are all race 1, and most are biovar 3 though some are biovar 4 (Hsu, 1991). These strains are all assigned to
1Corresponding author. Fax: 886-2-2902-1124; E-mail: bio1007@fujens.fju.edu.tw