Bot. Bull. Acad. Sin. (2001) 42: 159-166
Chow et al. Characterization of rice PAC P0699E04
Characterization of a 10 cM region of rice chromosome 5
Teh-Yuan Chow, Ya-Ting Chao, Su-Mei Liu, Hong-Pang Wu, Mu-Kuei Chu, Ching-San Chen, and Yue-Ie C. Hsing*
Institute of Botany, Academia Sinica, Taipei 115, Taiwan, Republic of China
(Received May 31, 2000; Accepted January 3, 2001)
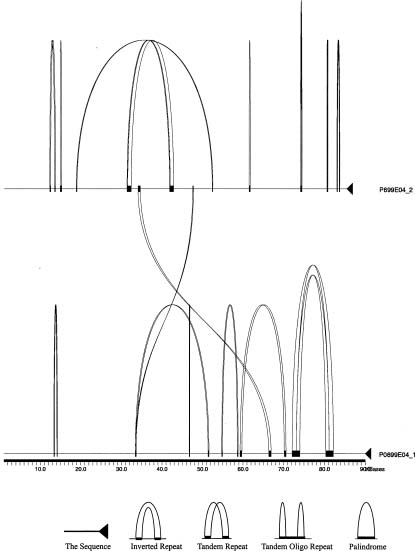
Abstract. Rice is a model species for the cereals and a good candidate for genome sequencing due to its relatively small genome (430 Mb), dense physical and genetic maps, and good transgenic systems. As part of an international effort to decode the rice genome, a PAC clone localized at 10 cM of chromosome 5 is completely determined for its sequence using shotgun libraries of its two inserts, 2-kb and 5-kb in length. In total 2,998 sequencing reads were used for the assembly of the final sequence, covering 175,439 bp. This sequence may code for at least 28 putative proteins, as deduced from computational search for homology with other known coding sequences and EST, or predicted using GenScan package. Also present in this sequence are simple repeats, palindrome and retrotransposons. On the basis of these findings, the gene density in the gene-rich region of rice genome is about 6 kb/gene.
Keywords: Annotation; High-throughput genome sequencing; Repetitive sequences; Retrotransposons; Rice genome.
Abbreviations: BAC, Bacterial artificial chromosome; EST, Expressed sequence tags; LTR, Long terminal repeat; NR, Non-redundant database; ORF, Open reading frame; PAC, P1-derived artificial chromosome.
Introduction
As the first genome of the higher plants, the small mustard species Arabidopsis thaliana will soon be completely sequenced. The sequences of chromosome 2 and 4 were recently published (Lin et al., 1999; Mayer et al., 1999). Additional knowledge of the genomes of other plant species is desirable to understand how plant genes evolved and are organized and regulated.
Rice (Oryza sativa) has been chosen as the first crop to be sequenced by an international sequencing consortium, the IRGSP (International Rice Genome Sequencing Project, Sasaki and Burr, 2000) for the following reasons: (1) Rice is an important crop in the world, feeding about one half of the world's population; (2) Rice's genome size, 430 Mb, is the smallest among crops (Arumuganathan and Earle, 1991); (3) Rice linkage and physical maps have been established (e.g. Harushima et al., 1998), and over 40,000 expressed sequence tags (ESTs) have been reported (Yamamoto and Sasaki, 1997) and mostly mapped. A yeast artificial chromosome (YAC) library that has been fingerprinted and ordered with mapped markers currently covers 60% of the rice genome (Kurata et al., 1997). Several bacterial artificial chromosome (BAC) libraries and P1-derived artificial chromosome (PAC) libraries have also been described. (4) The transgenic technology for rice has been established, and rice has become the easiest of all cereal plants to transform genetically. (5) Rice shares a co-linear gene organization with other cereal grasses and thus a key to knowledge of the genomic organization of the other grasses (Gale and Devos, 1998).
IRGSP, of which this lab is a member, adopts a map-based clone-by-clone shotgun strategy. Sheared bacterial artificial chromosome/P1-derived artificial chromosome (PAC) libraries are constructed from Oryza sativa ssp. japonica variety "Nipponbare" by labs in the States and Japan, respectively. BAC end-sequencing, fingerprinting and marker-aided PCR screening are used to make sequence-ready contigs. Using these libraries and information, each IRGSP member is for high-throughput sequencing and subsequent annotation of one or more of the twelve chromosomes. In this international effort, this lab in Taiwan works on the sequencing work of chromosome 5.
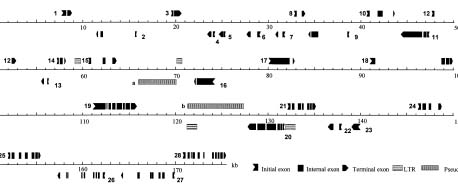
In this report, we describe the sequencing strategy and the annotation method used in the study. We also present the characterization of all the putative open reading frames and its repetitive sequence in P0699E04, a contig with a sequence localized around 10 cM of the short arm of rice chromosome 5.
Materials and Methods
Sequencing
P0699E04 is a PAC clone of the HindIII PAC library constructed by members of the Japan Rice Genome Research Program (RGP) using the genomic DNA of the japonica rice Nipponbare with the vector pCYPAC2. Its PAC DNA was sheared (1.6-2 kb and 4.5-5 kb), ligated to a pUC18 vector, and transformed into Escherichia coli. Sequencing reactions were performed using either BigDye Terminators, BigDye primers, or Dichlororhodamine Terminators (P.E. Biosystems) and were run on ABI377 sequencers (P.E. Biosystems). Shotgun clones were sequenced
*Corresponding author. Tel: 02-2789-9590 ext. 312; Fax: 02-2782-7954; E-mail: bohsing@ccvax.sinica.edu.tw