Bot. Bull. Acad. Sin. (2002) 43: 13-20
Chen et al. Sequencing and protein expression of TMMV
Molecular characterization of Tuberose mild mosaic virus and preparation of its antiserum to the coat protein expressed in bacteria
Chin-Chih Chen1, Tom Hsiang2, Fen-Lan Chiang1, and Chin-An Chang1,*
1Department of Plant Pathology, Taiwan Agricultural Research Institute, Wu-feng, Taichung 413, Taiwan, Republic of China
2Environmental Biology, University of Guelph, Guelph, Ontario, Canada
(Received April 3, 2001; Accepted September 12, 2001)
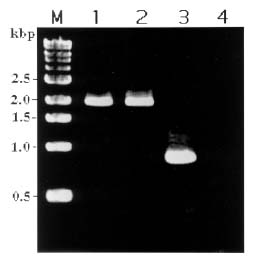
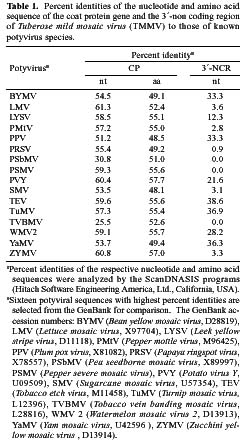
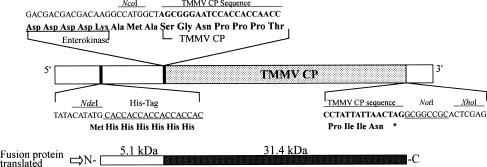
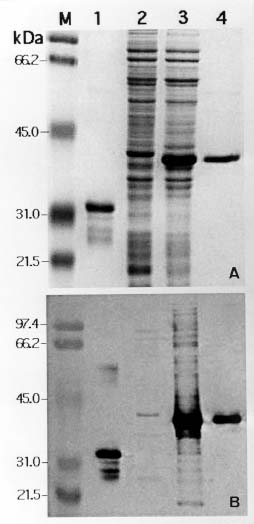
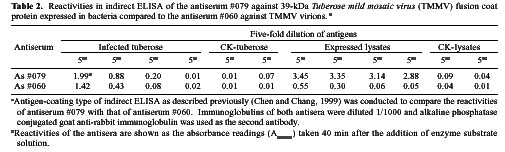
Abstract. A 2-kb DNA product was amplified from purified Tuberose mild mosaic virus (TMMV) virions as well as from infected tissues of tuberose by the use of degenerate primers for potyvirus. The PCR product was subsequently cloned and its sequence analyzed. It was found comprised of 1947 nucleotides (nts) corresponding to the 3´-terminal region of potyviruses. The deduced amino acid sequence contained 598 residues encoding part of the 3´-terminal region of NIb gene (319 residues) and the complete sequence of coat protein (CP) gene (279 residues). A 136 nts of non-coding region (NCR) was found located at the 3´-terminal region of the DNA. A genetic code for aphid transmissibility of potyviruses, DAG triplet, was found at the 19-21 residues from the N-terminus of CP gene. Compared to the known sequences of potyviruses, the percent of nucleotide identities of the CP gene and the NCR were less than 62% and 39%, respectively. Similarly, percent identities of TMMV's CP amino acid sequence to those of other known potyviruses were all below 58%, confirming our previous finding that TMMV is a new species of Potyvirus. Using directional cloning technology, a 39-kDa fusion protein containing a complete CP sequence of TMMV and a partial sequence encoded by the expression vector plasmid (pET-30b, Novagene) was highly expressed and purified from E. coli cell cultures. The antigenicity of the fusion protein was determined to be indistinguishable from the viral CP. Antiserum prepared against this fusion protein showed comparable reactivities in the serological detection of TMMV with the conventional antibodies against purified virus particles.
Keywords: 3´-terminal region; Antiserum; Expression vector; Fusion protein; Potyvirus; Sequences; Tuberose mild mosaic virus.
Introduction
Tuberose (Polianthes tuberosa L.) has been cultivated in Taiwan for over 300 years and has become an economically important ornamental bulb crop (Shen et al., 1987, 1993). Tuberose mild mosaic virus (TMMV), a newly recognized potyvirus, based on its serological and biological distinction to other known potyviruses, was recorded in 1998 (Chen and Chang, 1998). However, the molecular characteristics of this tuberose virus such as the coat protein (CP) gene sequence, the key taxonomic factor, have not been described (Chen and Chang, 1998). One of the major objectives of this study is to clone and analyze the sequences of the CP gene and the 3´-non coding region (3´-NCR) of TMMV. This sequence information is provided as molecular evidence for finalizing its actual taxonomic status. Secondly, due to generations of vegetative propagation, all of the major tuberose varieties in Taiwan have been found infected by TMMV. Preparation of specific antiserum was hampered by the difficulties in obtain
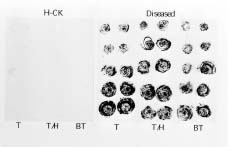
ing sufficient quantities of purified TMMV (Horner and Person, 1988). Recently an antiserum against TMMV was successfully prepared and applied in virus identification and indexing in Taiwan (Chen and Chang, 1998). Using the antiserum, a complete virus indexing and bulb certification program for tuberose has been developed (Chen et al., 1998; Chen and Chang, 1999). In addition, the clone of a virus-free multi-petal tuberose variety was rescued by meristem-tip tissue culture and identified by indexing with this antiserum (Chen et al., 1999). As a result, an industrialized virus-free tuberose bulb propagation program is becoming established. However, its success will depend upon a consistent supply of antiserum to TMMV. The efficient production of good quality antiserum to TMMV is not an easy task under conventional methods due to their instability and extremely low yield in tuberose tissue. In this study, we took the approach of cloning the complete coat protein (CP) gene and expressing it in a bacterial culture system. The bacteria-expressed viral CP was then efficiently purified in sufficient quantity for antiserum preparation. Serological experiments showed that antiserum prepared by this approach was equivalent to traditional antiserum against purified virions in the detection of TMMV in tuberose tissue.
*Corresponding author. Tel: 886-4-23302803; Fax: 886-4-23331089; E-mail: cachang@wufeng.tari.gov.tw