Bot. Bull. Acad. Sin. (2004) 45: 275-283
Chen et al. b-N-acetylhexosaminidases from mungbean seedlings
Purification and characterization of isoforms of b-N-acetylhexosaminidase from mungbean seedlings
Yi-Ching CHEN1,6, Wei-Liang LIU2,6, Hui-Ching HSU3, Yung-An LEE2, and Ching-San CHEN3,4,5,*
1Graduate Institute of Bioscience and Biotechnology, National Ocean University, Keelung 202, Taiwan
2Department of Life Science, Fu-Jen Catholic University, HsinChuang, Taipei 242, Taiwan
3Institute of Botany, Academia Sinica, Nankang, Taipei 115, Taiwan
4Department of Agricultural Chemistry, National Taiwan University, Taipei 106, Taiwan
5Institute of Microbiology and Biochemistry, National Taiwan University, Taipei 106, Taiwan
(Received June 2, 2004; Accepted July 19, 2004)
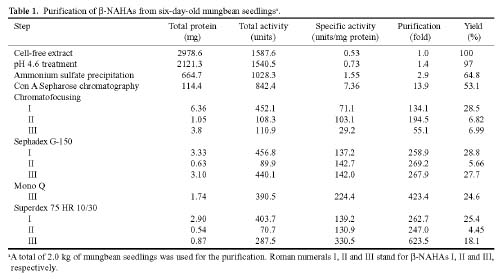
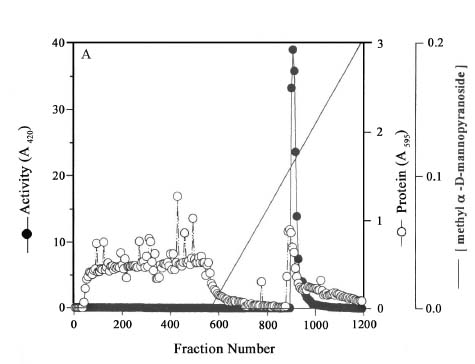
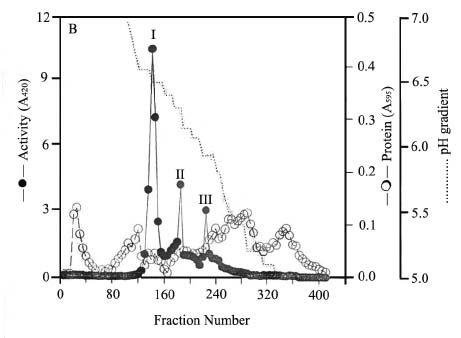
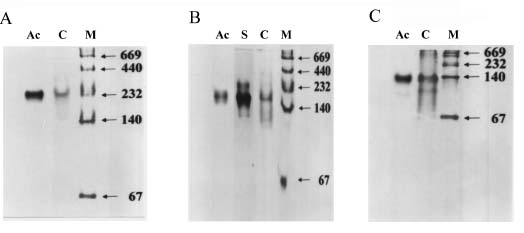
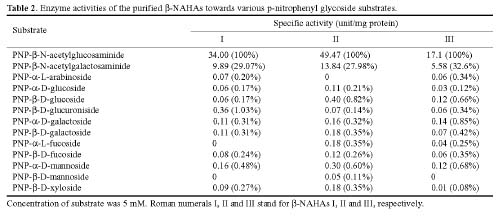
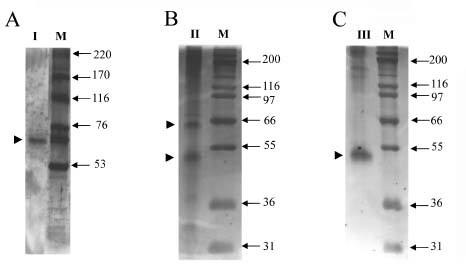
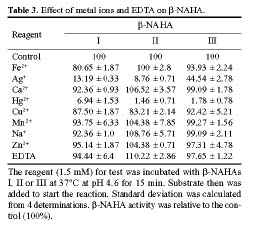
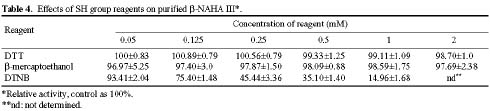
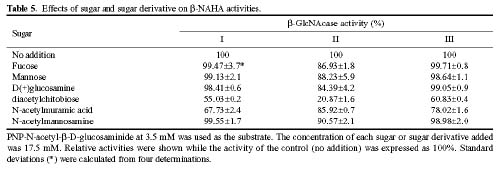
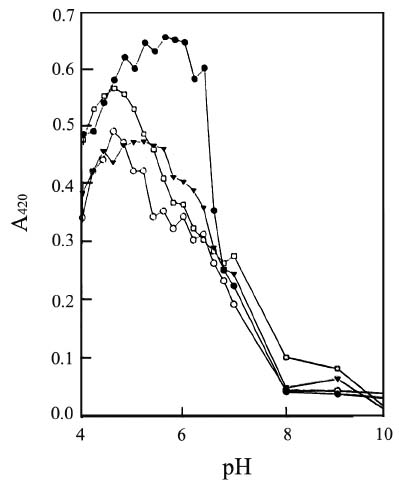
Abstract. Three isoforms of b-N-acetylhexosaminidase (b-NAHA), named b-NAHAs I, II and III, were isolated from six-day-old etiolated mungbean (Vigna radiata) seedlings. b-NAHA I was purified to apparent homogeneity by a procedure involving Con A-Sepharose chromatography, chromatofocusing, and gel filtration. b-NAHAs II and III were highly purified. b-NAHAs I, II and III had molecular masses of 135,127 and 110 kDa, respectively. b-NAHA I was dissociated into a single 67 kDa protein band. II was dissociated into two protein bands corresponding to 60 and 48 kDa, and III was dissociated into a single 48 kDa protein band in SDS-polyacrylamide gel electrophoresis. The results suggest that isoforms I and III are homodimeric enzymes, each comprising two identical subunits with molecular masses of 67 kDa and 48 kDa, respectively, while isoform II is a heterodimeric enzyme, comprising two non-identical subunits with molecular masses of 60 kDa and 48 kDa. All the enzymes were active against para- nitrophenyl-b-N-acetylglucosaminide (PNP-b-N-acetylglucosaminide) and PNP-b-N-galactosaminide. The enzymes were inhibited by 5, 5'-dithiobis (2-nitrobenzoic acid)(DTNB), Ag+, Hg2+, and N, N'-diacetylchitobiose. Km values for isoforms I, II and III were 0.67 mM, 1.04 mM and 1.76 mM, respectively, using PNP-b-N-acetylglucosaminide as a substrate. These three isoforms had acidic pI values (I, 6.3; II, 6.1; and III, 5.9). Their optimal pH in the reaction towards PNP-b-N-acetylglucosaminide was 5.4, 4.7 and 5.7, and optimal temperatures were 65°C, 65°C and 50°C for isoforms I, II and III, respectively.
Keywords: b-N-acetylhexosaminidase; Enzyme purification; Germination; Oligomeric structure; Vigna radiata.
Abbreviations: DTNB, 5,5'-dithiobis-2-nitrobenzoic acid; DTT, dithiothreitol; b-GlcNAcase, N-acetyl-b-D-glucosaminidase; b-GalNAcase, N-acetyl-b-D-galactosaminidase; b-NAHA, N-acetyl-b-D-hexosaminidase; PMSF, phenylmethylsulphonyl fluoride; PNP, p-nitrophenyl.
Introduction
b-N-Acetylglucosaminidase (b-GlcNAcase) (EC3,2,1,30) catalyzes the release of N-aetylglucosaminyl residue from the non-reducing terminus of oligosaccharides. It is also referred to as b-N-acetylhexosaminidase (b-NAHA) because it can also cleave terminal N-acetyl-b-D-galactosaminyl residues from oligosaccharides (Dey and Campillo, 1984; Conzelmann and Sandhoff, 1987). This enzyme occurs widely in animals, microorganisms, and plants and has been implicated in a number of biological processes, including the degradation of glycoproteins (Bahl and Agrawal, 1968; Li and Li, 1970; Neely and Beevers, 1980; Poulton et al., 1985), glycolipids (Fernandes et al., 1997) , and N-glycans (Choi and Gross, 1994). In
plants, b-NAHA is thought to play a vital role during seed germination by cleaving N-acetylglucosaminyl residues from storage glycoproteins which have undergone prior proteolytic degradation. The liberated amino sugars and amino acids are then available for further synthesis of glycoproteins in the developing seedlings. This has been supported by the finding that b-NAHA activities increase during the process of germination (Bahl and Agrawal, 1968; Bouquelet and Spik, 1978; Yi, 1981). A prominent increase in b-NAHA activity was found in the germinating seeds of Phaseolus vulgaris. The specific activity increased 25-fold upon germination (Bahl and Agrawal, 1968). The enzyme may also have a role in the posttranslational trimming of oligosaccharide chains of the storage proteins and lectins deposited in the protein bodies during seed development (Vitale and Chrispeels, 1984). Activities of b-NAHA and chitinase were demonstrated in whole tissues of individual seeds and seedlings using an in situ technique (Hodge et al., 1996). It is believed that chitinase acts as a defense protein in higher plants, protecting the plant from
6Y.C. Chen and W.L. Liu contributed equally to this paper.
*Corresponding author. Phone: +886-2-27899590 ext 251; Fax: +886-2-27858936; E-mail: chingsan@gate.sinica.edu.tw