392
Botanical Studies, Vol. 50, 2009
dNTPs, 1 fil diluted ligation product, 0.5 jiM of each E00 and H/M00 primers and 1 unit Taq polymerase (TaKaRa, Japan). These products were also diluted 10-fold with sterile water, and used as the template for selective amplification. Selective amplifications were conducted in a final volume of 20 fil, containing 1 fil of the diluted pre-amplification product and 0.5 jiM each of E and H/M primers; the remaining components were the same as those in the pre-amplification reaction. The reaction was carried out as described by Xiong et al. (1999). The denatured PCR products were separated on a 6% denaturing polyacrylamide gel at 35 W for 2 h. The gel was then stained with silver (Chalhoub et al., 1997). Only clear and reproducible bands that appeared in two independent PCR amplifications [starting from the DNA digestion step, (i.e., the first step of MSAP) were scored].
RESULTS
Validation of the ploidy level by Karyotype assay
The ploidy levels of all materials used in present study were determined by counting chromosome numbers in root
cells. A total of ten individuals randomly selected from each of three different ploidy watermelons were tested. At least eight cells from two independent root tips of each sample were used to count the number of chromosomes. The results indicated that in each cell, the corresponding chromosome numbers were completely consistent with previous reports (Beevy and Kuriachan, 1996), implying that all materials were stable with respect to ploidy level, and no significant changes in chromosome structure had occurred (Figure 1).
Alteration in genomic structure detected by ISSR and SRAP assays
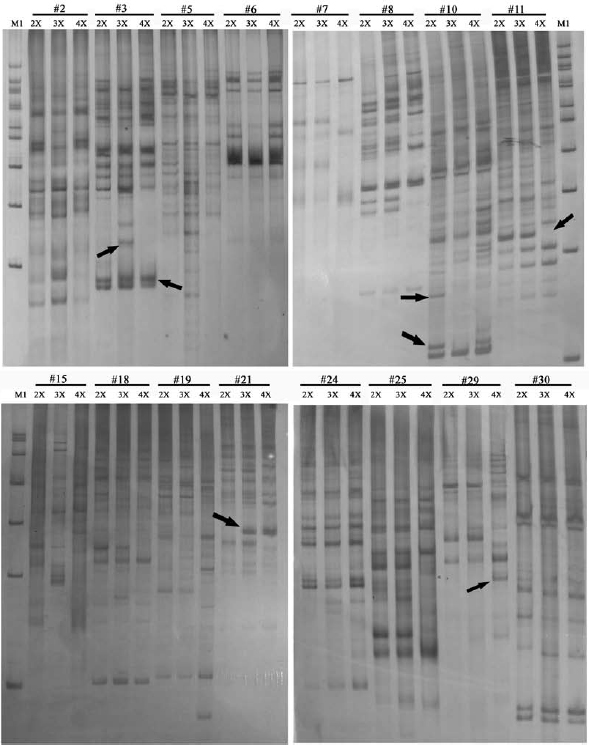
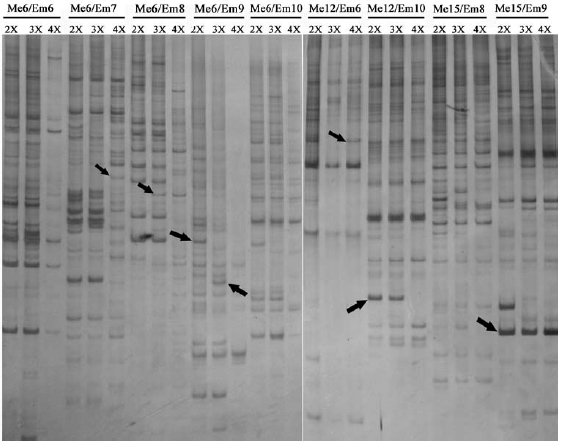
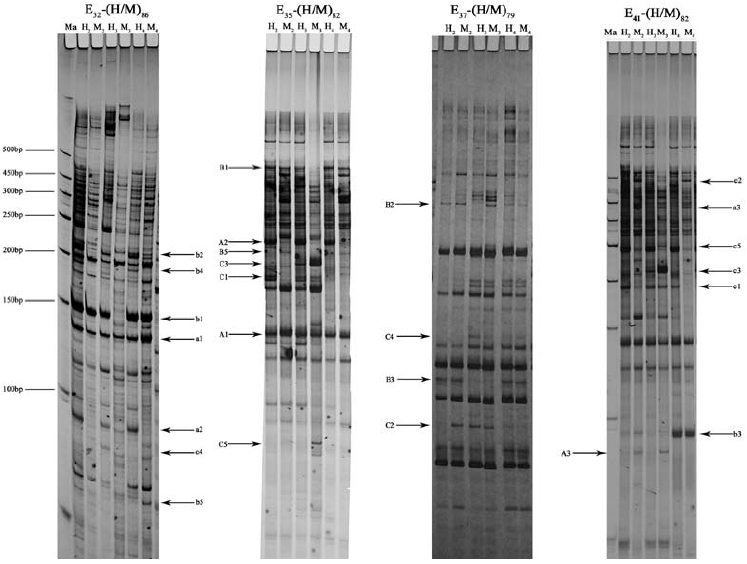
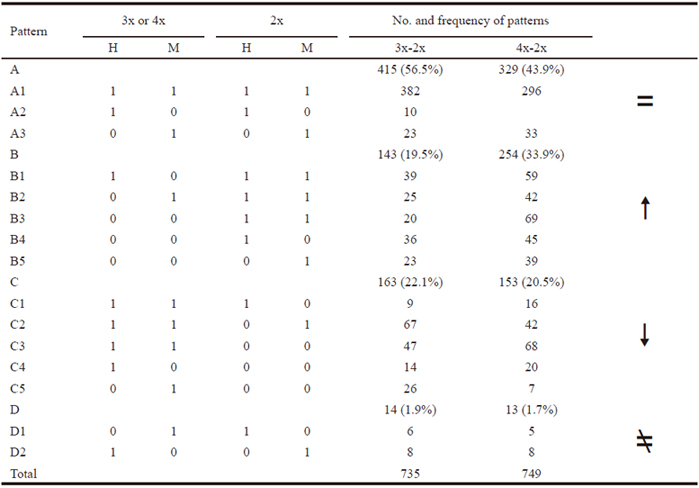
The ISSR assay generated approximately 1000 scored bands, for an average of 333 bands per ploidy sample, representing 328 prospective genetic loci. Partial results are presented in Figure 2. Among these 328 loci, 182 sites were simultaneously detected in three different ploidy samples, meaning that the remaining 146 were polymorphic, and the polymorphism frequency was thus 44.5% (146/328). A comparison of sites detected in 3x with those in 2x, showed that 17.4% (57/328) of sites were changed, of which 7.6% (25/328) sites were specifically

Figure 1. Karyotype of different ploidy watermelon. (a) diploid watermelon, JKR-1 (2n=2x=22); (b) autotriploid watermelon, JKR-2 (2n=3x=33); (c) autotetraploid watermelon, JKR-3 (2n=4x=44); Scale bar=5 (im.