406
Botanical Studies, Vol. 50, 2009
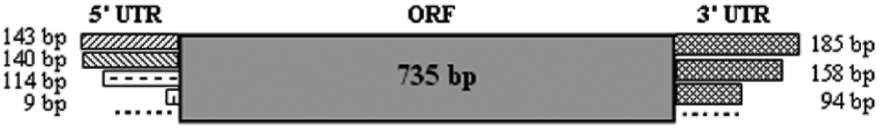
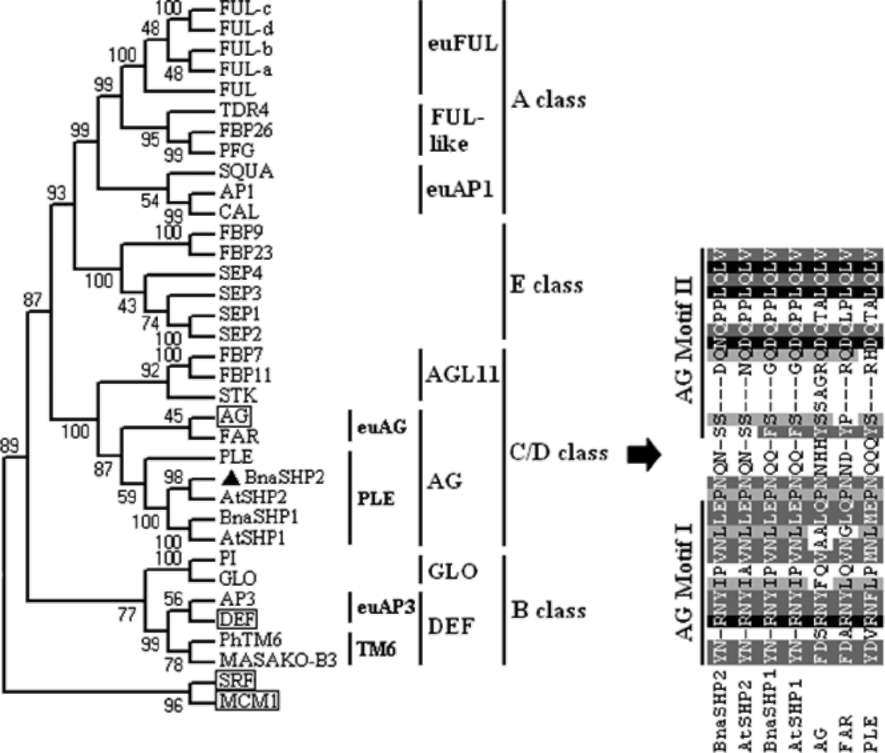
Figure 1. Schematic diagram of different transcripts of BnaSHP2 obtained by RACE. The diverse 5' UTR, four cDNA sequences of which were sequenced, were described in front of ORF region, while three different lengths of 3' URT were scaled out on the right of Figure. The possible other sequences of 5' URT and 3' URT were indicated with "......
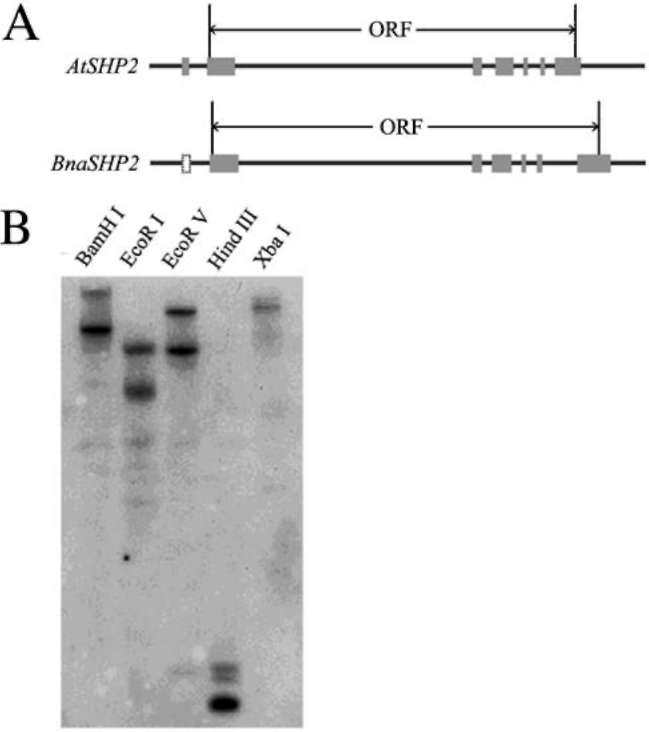
sequenced, while other possible sequences are still not known. It is interesting that these four 5'-UTR sequences are quite divergent and three 3' -URT sequences are identical. According to these sequences, four upstream primers and three downstream primers were designed and used to amplify the full length of cDNA of BnaSHP2 by their cross-pairing, so several PCR products ranged from 838 bp to 1,063 bp were obtained, indicating that multiple transcripts existing in BnaSHP2. These transcripts shared an almost same ORF (735 bp) sequence except for fifteen nucleotides, and were divided into two groups (a and p). This may suggest that these transcripts of BnaSHP2 may be derived from two paralogues. In addition, four genomic DNA clones from B. rapa (GenBank AC189588 and DU113592), B. oleracea (BZ061644) and B. napus
(FP018823) are found in TAIR database of genomic DNA.
The genomic DNA clones from B. rapa and B. napus are consistent with group a while that from B. oleracea is consistent with group p, indicating that transcripts in group a are derived from A genome while those in group p are derived from C genome. According to the standardization of Brassica gene naming provided by 0stergaard and King (0stergaard and King, 2008), core sequences of two groups represent two genome types of BnaSHP2 are named respectively as BnaA.SHP2.a and BnaC.SHP2.b
(GenBank Acc. No. EU424342 and EU424343). Their
putative amino acid sequences of two paralogues of BnaSHP2 are almost identical except for one amino acid residue (S or N) at the position of 187aa (Figure 2). In this paper, we used BnaA.SHP2.a for subsequent analysis and experiments (if without specification).
Expasy tool (http://www.expasy.org/) was used to analyze profiles of the BnaSHP2 protein. The 735 bp ORF of BnaSHP2 encoded a protein of 244 amino acids with a predicted molecular weight of 28 kDa and a calculated isoelectric point of 9.12. Total number of negatively charged residues (Asp + Glu) and positively charged residues (Arg + Lys) are 31 and 37, respectively. The
BnaSHP2 has three N-glycosylation sites (28 to 31 NTTN) (74 to 77 NNSV) (232 to 235 NSSD), eight Protein kinase C phosphorylation site (12 to 14 SSK) (13 to 15 SKK) (30 to 32 TNR) (64 to 66 STR) (76 to 78 SVR) (109 to 111 SSK) (155 to 157 SKK) (187 to 189 SER), two casein kinase II phosphorylation site (96 to 99 SVTE) (205 to
208 TVYE).
Structure analysis of BnaSHP2 and homologous alignment
Using SMART program (Simple Modular Architec-
at ggaggft ggt gc gagt gat^J^t age ag&gagc age aagaagat ag^gagagg^ag M £ G G A S D F^V AESSKKI&RGK
2D 40
60
SO
100
120
140
160 130
200
220 240
~t-it
h b 0 v
ni. ggt c tgc JtLftgft&^^c tt at gage tctct gti ttgtgt ^.c tgag^t tgetett HGLLKKAYELSVLCBitEVAL
VIFSTRGRLYEY AHHEVRGT c
at t gaa&g^t ac ftftga&^^c ttgetec gac t gtt &At cetccttc cgt cacc ga&gc t IERTKKACSEAVHFFSVTEA
ait actc ac t ate a^c aag^t c^j- ctaagc tac ggagacaeat cc eee^c ftttcae NTQYYQQESSKLRJtQIRDIQ
9LHR»ILG£SLGSLNLK EL K
aac c tQ^aa^gt aggc 11 gJ^aaggc " c ggt cgtgtccgctcc aagaagc " gag" g K L E G R L E^K GIGRVKSKKHHM
tatc tluc c c aagat t satgaaa gc aggaa t gc age agcaggaagc gagt gt a TLRSKISEKAGMQQQEXSVI H
A R
HQQGTVYE S SHQSZQYNItN TIFVILLEFMONSSDQHQFF
L S L v - 244
Figure 2. Core cDNA sequence of the BnaSHP2 gene and deduced amino acid sequence from it the core nucleotide sequence of BnaA.SHP2.a is shown, and only the nucleotides in BnaC.SHP2.b that are different from BnaA.SHP2.a are shown in little boxes upon the BnaA.SHP2.a sequence. The amino acid sequence of BnaA.SHP2.a was shown beneath the nucleotide sequence. "―"indicates the different amino acid residue between BnaA.SHP2.a and BnaC.SHP2.b. MADS-box domain signature region of BnaSHP2 is underlined. A bipartite nuclear localization signal was predicted and marked with rectangular box.
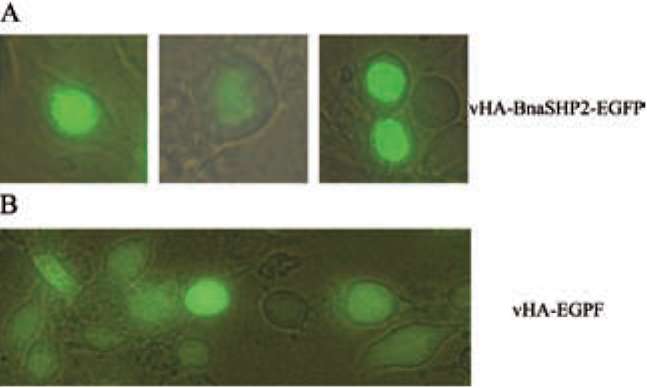
ture Research Tool: http://smart.embl-heidelberg.de), A MADS-box domain (amino acid 18-72) and a K-box domain (amino acid 102-192) as DNA-binding and dimer-ization regions were identified in the BnaSHP2, indicating that the protein belongs to MADS-box transcription factor family. By comparing BnaSHP2 with other MADS-box proteins, the conservative sequence was primarily restricted to MADS-box and K-box regions. Other regions showed a high degree of variation among the MADS-box proteins compared (data not show). Sequence alignment of the MADS-box and K-box domains from BnaSHP2 and related MADS-box proteins from yeasts, plants, and humans indicated that the MADS-box is the most conservative region in all MADS-box members' proteins,