SUI et al. ― cDNA library construction and ESTs analysis of B. chinense
9
results
Overall features of B. chinense root full-length enriched cDNA library
One microlitre of transformed ligation mixture yielded 1113 recombinant clones, and the titre of the full-length enriched cDNA library was about 1.1x106 clones. To evaluate the size and distribution of insert cDNA clones, a total of 136 clones were randomly selected and digested by Sfil, allowing us to obtain average cDNA insert sizes and the cDNA length distribution profiles. Most insert lengths ranged from 0.5 to 2 kb. The estimated average insert size was 1.1 kb. The size distribution of insert DNA clones is shown in Figure 1.
A total of 3902 recombinant clones were randomly selected and sequenced from the 5' end. After eliminating low-quality sequences and contaminated clones, 3111 valid sequences in all were obtained. Clustering and assembly of these ESTs resulted in a total of 1650 uniESTs with 377 contigs and 1273 singleton ESTs. BlastX and BlastN analysis showed that of 1650 uniESTs, 949 (57.5%) were homologous to previously identified genes, 680 (41.2%) matched to unknown, unnamed, or hypothetical protein genes, and 21 clones had no hit. Clones corresponding to the identified genes were tested to determine whether they contained a 5' UTR and a putative ATG translation initiation codon. Among these sequences matched to a known gene, 489 (ca. 51.5%) clones were predicted to contain a putative ATG translation initiation codon, and among them 159 single sequences were deposited in the dbEST division of GenBank (Accession numbers: FG341847-FG342005). General features of the library and sequencing statistics are listed in Table 1.
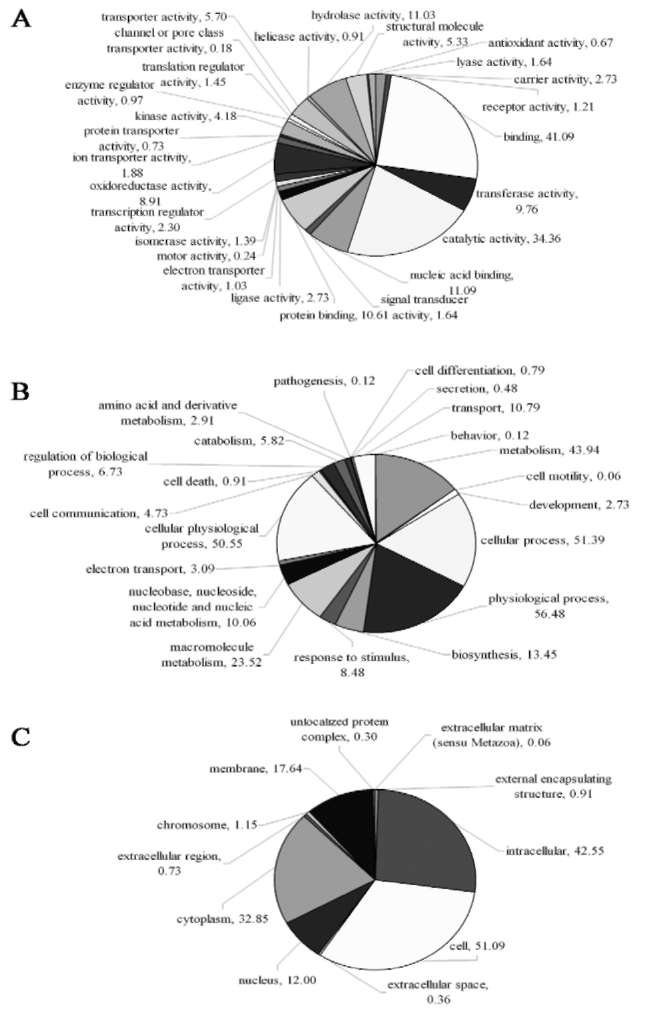
Gene Ontology annotation and metabolism pathway analysis
All uniESTs were assigned the Gene Ontology (GO) terms using the sequence comparison results of BlastX. Of the uniESTs, 1002 (60.7%), 957 (58.0%), and 861 (52.2%) were assigned molecular function, biological process, and cellular component GO terms, respectively. One uniEST did not exclusively belong to a single GO term. Figure 2 illustrates the GO term distribution of all uniESTs. The first two largest categories in three GO terms were binding and catalytic activity, physiological process and cellular process, cell and intracellular. These results which relate to the storage function of the root were consistent with similar studies on P. ginseng root (Jung et al., 2003). Comparison with the KEGG Arabidopsis pathway showed that 307 uniESTs represented 31 metabolic pathways on the prerequisite that each pathway contained at least 5 uniESTs (Table 2). The full-length enriched cDNA library we constructed is thus useful in identifying functional genes of B. chinense. BlastN analysis of our uniESTs with Bupleurum nucleotide sequences obtained from GenBank indicated that only three were homologous and the hits were AM409304 (cinnamic acid 4-hydroxylase),
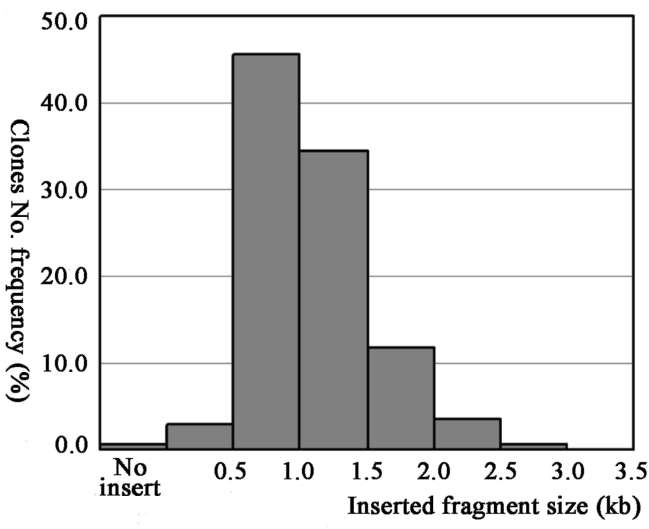
Figure 1. Size distribution of cDNAs in the library. Fragment sizes were determined by sfil digestion of 136 random selected clones.
Table 1. General features of root of B. chinense full-length enriched cDNA library and sequencing statistics.
Titre (pfu)
Average cDNA insert size Total clones sequenced Sequences passed qualiy check Clustered (contigs) Unassembled (singletons) Unigenes (contigs+ singletons) Observed redundancy"1 No hit to nr (BLASTX)
1.1x106 1.1 kb 3902 3111 (79.7%) 377 1273 1650 88.5%
22
EST matches with E-value in BLASTX > 1><10—14 366 (11.8%) EST matches with E-value in BLASTX < 1><10—14 2724 (88.2%)
aOberved redundancy: (EST# after quality check-Unigene #)/ Unigene # (Lindqvist et al., 2006).
AM409290 (short-chain dehydrogenase/reductase), and AM409292 (omega-6 fatty acid desaturase). Therefore, nearly all uniESTs that we sequenced may represent novel transcripts from Bupleurum.
To visualize the transcript abundance of Bupleurum root, the contigs assembled from the library were analyzed. Of these, the sixteen most abundant transcripts (EST number equal to or larger than 18) observed in this library are listed in Table 3. The contig with the largest number of ESTs was identified to be dehydrin protein. Contigs with the second and third largest ESTs were a pathogenesis-related protein-like protein 1 and a putative