Botanical Studies (2010) 51: 511-520.

Reexamination of the pattern of geographical disjunction
of Chamaecyparis (Cupressaceae) in North America and
East Asia
of Chamaecyparis (Cupressaceae) in North America and
East Asia
Pei-Chun LIAO1,2, Tsan-Piao LIN3, and Shih-Ying HWANG1*
1 Department of Life Science, National Taiwan Normal University, 88 Ting-Chow Road, Section 4, Taipei 11677, Taiwan
2Department of Life Science, National Pingtung University of Science and Technology, 1 Shuefu Road, Neipu, Pingtung 912, Taiiwan
2Department of Life Science, National Pingtung University of Science and Technology, 1 Shuefu Road, Neipu, Pingtung 912, Taiiwan
3Institute of Plant Biology, National Taiwan University, 1 Roosevelt Road, Section 4, Taipei 10617, Taiwan
(Received November 24, 2008; Accepted March 17, 2010)
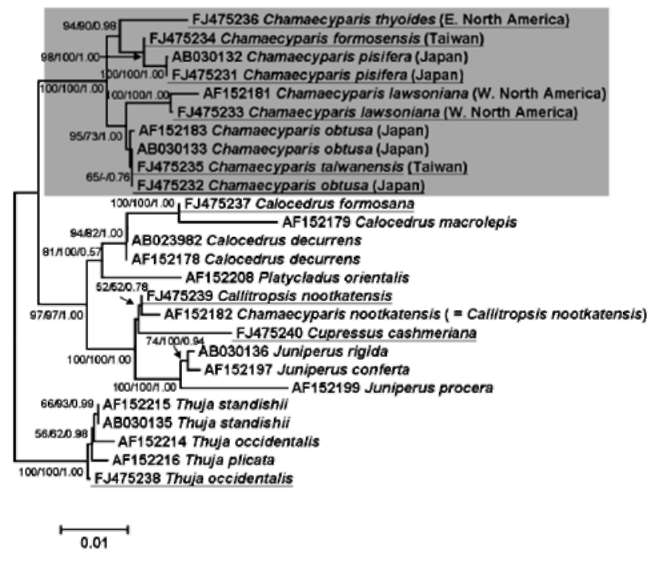
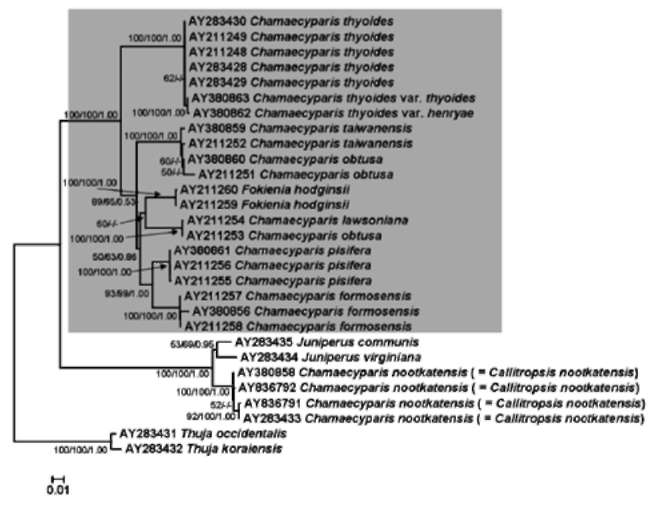
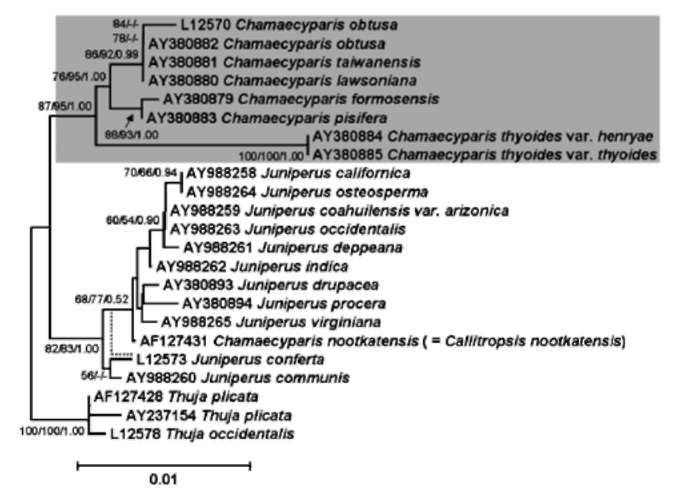
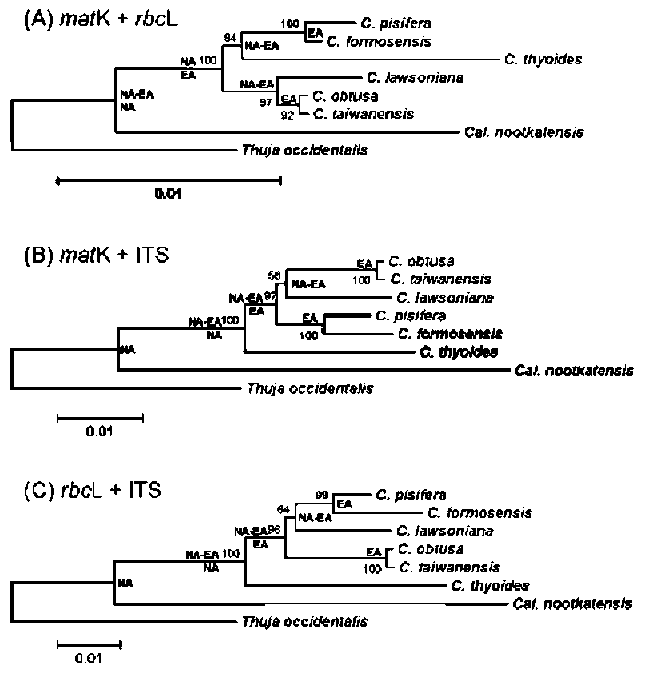
ABSTRACT. The genus Chamaecyparis (Cupressaceae) is comprised of six taxa native to East Asia and North America. Two independent lineages from eastern North America to East Asia and from western North America to East Asia have been identified from phylogenetic analyses based on variation in plastid DNA (ptDNA). However, the trees inferred from a nuclear ribosomal internal transcribed spacer (nrITS), the NEEDLY intron 2, and combined data from genetic and morphological characters of other studies are incongruent with the tree inferred from ptDNA. In this study, we sequenced the plastid matK gene as well as other sequences acquired from GenBank including the matK, rbcL, and nrlTS sequences to reconstruct the phylogenetic trees of Chamaecyparis. The phylogenetic topology inferred from matK was congruent with the previous inference obtained using other ptDNA markers while the rbcL and ITS trees were congruent with previously inferred trees using the combined data and ITS sequences, respectively. Relatively less- (rbcL) and more-informative sites (ITS) may lead to different lineage sorting and incongruent phylogenetic topologies, which were evidenced by rejection of evolutionary homogeneity between ptDNA and ITS sequences in the partition homogeneity test. The phylogenetic tree reconstructed using matK and other ptDNA strongly supports the geographically disjunct distribution of Chamaecyparis in North America and East Asia. A dispersal-vicariance analysis, and geologic and fossil evidence indicated that at least two independent dispersal events occurred from North America to East Asia, which support the previous biogeographic inference by ptDNA. The use of biparental inherited markers for biogeographic inferences should be done with caution.
Keywords: Chamaecyparis; Geographical disjunction; Phylogenetic relationships; matK.
INTRODUCTION
Chamaecyparis is a genus with a fragmented distribution in East Asia and North America. The genus Chamaecyparis is comprised of six taxa: the Japanese endemic species C. obtusa (Siebold & Zucc.) Endl. and C. pisifera (Siebold & Zucc.) Endl., the Taiwanese endemic species C. taiwanensis (C. obtusa Siebold & Zucc. var. formosana (Hayata) Hayata) and C. formosensis Matsum., the eastern North America endemic species C. thyoides (L.) Britten, Stern & Poggenb., and the western North America endemic species C. lawsoniana (A. Murray bis) Pari. Chamaecyparis nootkatensis (D. Don) Spach, formerly classified as a species of Chamaecyparis, was suggested to be removed from Chamaecyparis and
*Corresponding author: E-mail: hsy9347@ntnu.edu.tw; Tel: +886-2-29326234 ext. 221; Fax: +886-2-29312904.
recognized as belonging to the genus Cupressus by Welch (1991) and Frankis (1993), but it is now named Callitropsis nootkatensis (D. Don) Orest. (Little et al., 2004).
Previous phylogenetic inferences based on plastid DNA (ptDNA) segments [i.e. petG-trnP intergenic spacer and trnV intron (Wang et al., 2003) and matK, trnL, and rbcL sequences (Little, 2006)] supported two monophyletic groups of Chamaecyparis consisting of 1) C. lawsoniana, C. obtusa, and C. taiwanensis, and 2) C. thyoides, C. pisifera, and C. formosensis. According to these inferred phylogenetic relationships, there is a biogeographic pattern of two long-distance dispersals from eastern and western North America to East Asia (Japan and Taiwan). These events were proposed to have occurred from the middle to late Miocene (14 and 5.5 million years ago (mya), respectively; (Wang et al., 2003). However, incongruent phylogenetic relationships were inferred using the nuclear