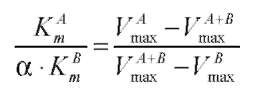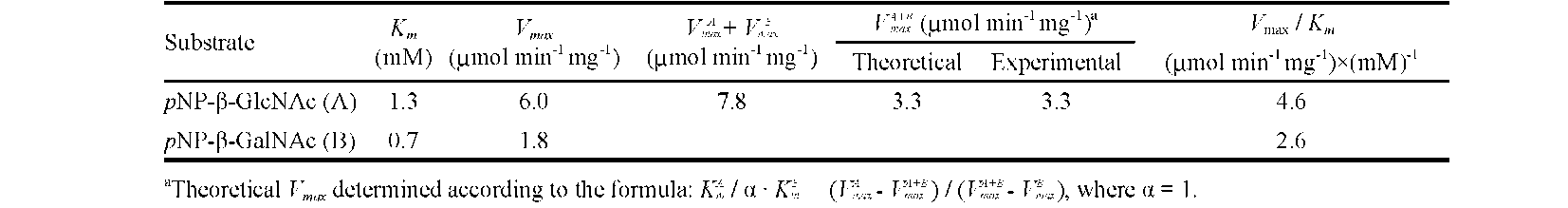Botanical Studies (2011) 52: 23-34.

Biochemical characterization of a β-N-acetylhex-osaminidase from fig latex
Ya-Min CHANG1, Yun-Chin CHUNG1, Chia-Chen HSU1, Li-Chun CHEN1, Chui-Liang CHIANG2, Chen-Tien CHANG1*, and Hsien-Yi SUNG3'*
1Department of Food and Nutrition, Providence University, Shalu, Taichung 433, Taiwan
2Department of Food Science, Central Taiwan University of SScience and Technology, Taichung 406, Taiwan
3Department of Biochemical Science and Technology, National Taiwan University, Taipei 106, Taiwan
(Received April 16, 2010; Accepted July 8, 2010)
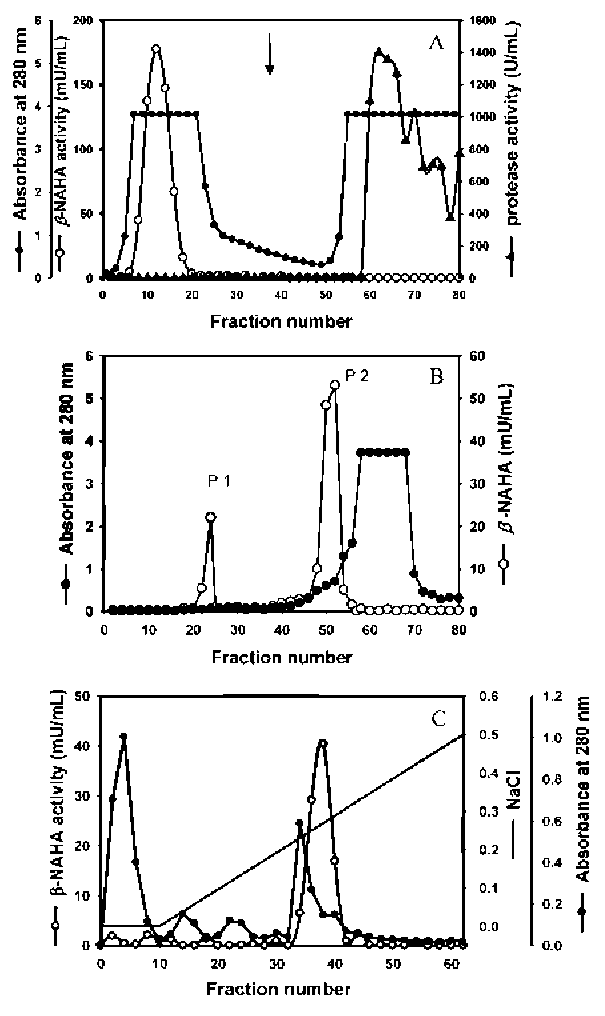
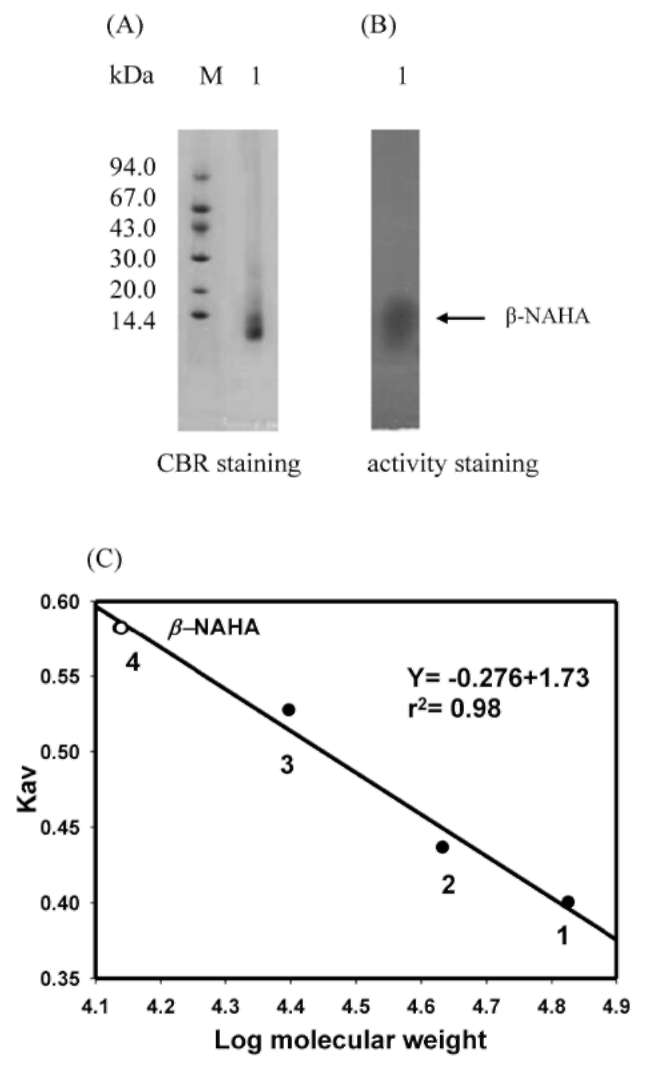
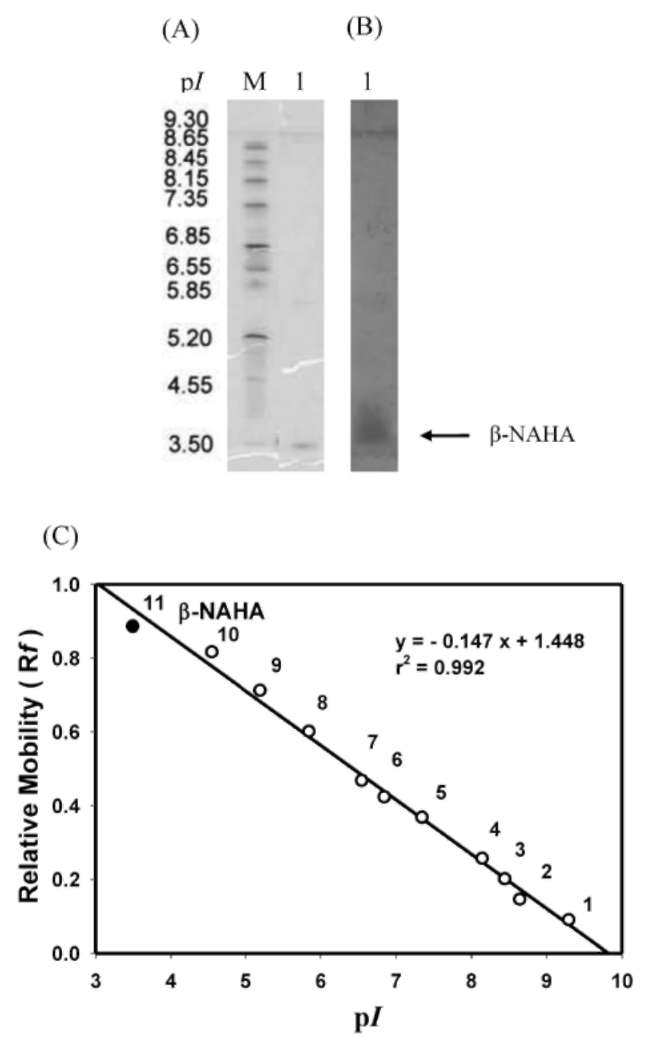
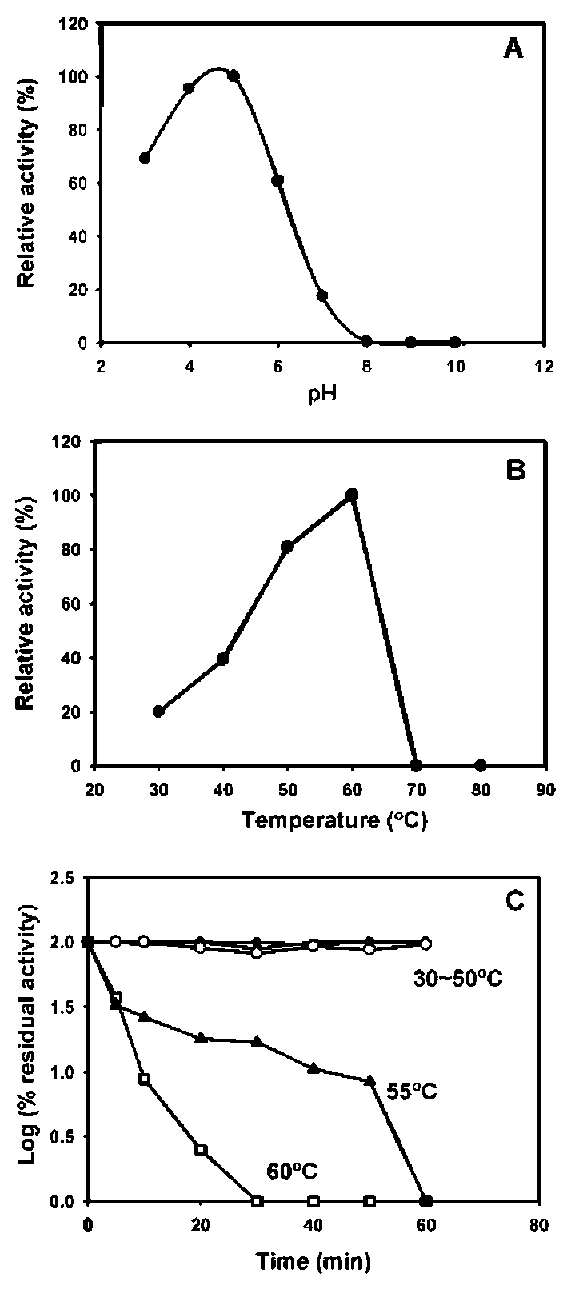
ABSTRACT. A major isoform of β-N-acetylhexosaminidase (β-NAHA) (EC 3.2.1.52) was purified from fig latex in three chromatography steps, affinity chromatography on pAPMA-Sepharose CL-4B to remove cysteine proteases, Sephacryl S-100 HR gel filtration, and DEAE-Sephacel ion-exchange chromatography. The purified β-NAHA appeared almost homogeneous on SDS-polyacrylamide gel electrophoresis and enzyme-activity staining. The purified enzyme catalyzed the hydrolysis of both p-nitrophenyl-N-acetyl-p-D-glucosaminide (pNP-β-GlcNAc) and β-nitrophenyl-N-acetyl-β-D-galactosaminide pNP-β-GalNAc). The optimum pH for the pNP-β-GlcNAc hydrolysis was 4.5, the optimum temperature was 60°C, the Km was 1.3 mM, the Vmax was 6.0 fimol min-1 mg-1 and the activation energy was 8.93 kcal/mol. The molecular mass of the enzyme was 13.7 kDa, as estimated by gel filtration. The isoelectric point of the enzyme was 3.5, as estimated by isoelectric focusing electrophoresis and activity staining. The enzyme was thermally stable after 60 min at 30-50°C, but its activity decreased significantly at temperatures greater than 55°C. Both the heavy metal ion Hg2+ (0.25 mM) and the chemical modification reagent diethyl pyrocarbonate (2.5 mM) significantly inhibited enzyme activity. Substrate specificity and competition kinetics analysis indicated that the activity of the purified β-NAHA was specific for the p-glycosidic linkage, and the enzyme had only one active site for substrates, pNP-β-GlcNAc and pNP-β-GalNAc.
Keywords: Characterization; Fig (Ficus carica) latex; β-N-acetylhexosaminidase; Purification.
INTRODUCTION
also seen in the whole tissues of individual seeds and seedlings (Hodge et al., 1996) and in rubber tree latex (Martin, 1991; Giordani et al., 1992). Some plant β-NAHAs also degrade chitin and chitin oligomers (Li and Li, 1970; Yi, 1981; Barber and Ride, 1989), suggesting that the enzyme plays a role in the defense system in plants against chitin-ous pathogens. However, the physiological signification of this enzyme in plants has yet to be fully elucidated.
Latex is the cytoplasmic fluid, containing the usual plant organelles in laticifer cells. Laticifers are anastomosed as a result of partial hydrolysis of adjacent walls, and thus form a tube-like network, or paracirculatory system, through-out the plant (Esau, 1967). When laticifers are injured, latex flows from the wound site. The latex exuded from laticifers is known to contain a variety of defense-related proteins such as chitinase, p-1,3-glucanase, hevamine and hevein (Broekaert et al., 1990; Rozeboom et al, 1990; Van Parijs et al., 1991). It has been suggested that latex secretion provides a defense against wounds and/or predators such as insects and microorganisms. Fig tree latex contains proteolytic enzymes (ficin) (Robbins and Lamson, 1934; William, 1958; Whitaker, 1959), chi-tinolytic enzymes such as plant lysozyme (Glazer et al.,
β-N-acetylglucosaminidase catalyzes the release of N-acetylglucosaminyl residues from the non-reducing terminus of oligosaccharides. It is also referred to as p-N-acetylhexosaminidase (β-NAHA) because it is capable of cleaving terminal N-acetyl-β-D-galactosaminyl residues from oligosaccharides as well (Dey and Campillo, 1984; Conzelmann and Sandhoff, 1987; Webb, 1992). This enzyme is widely distributed in nature and has been detected in animal tissues, microorganisms, and plants (Conzel-mann and Sandhoff, 1987). In plants, β-NAHA is thought to participate in the processing and turnover of glycopro-tein during germination (Neely and Beevers, 1980; Yi, 1981; Vitale and Chrispeels, 1984) and the metabolism of N-glycans during ripening in apples (Choi and Gross, 1994). However, β-NAHA and chitinase activites were
*Corresponding authors: E-mail: sunghy@ntu.edu.tw; Fax: 886-2-23634729; Tel: 886-2-33664522 (Hsien-Yi SUNG); E-mail: ctchang@pu.edu.tw; Fax: 886-4-26530027; Tel: 886-4-26530089 (Chen-Tien CHANG).