Bot. Bull. Acad. Sin. (2001) 42: 23-33
Yuan et al. Glutamine synthetase from Scynechococcus RF-1
Purification and characterization of glutamine synthetase from the unicellular cyanobacterium Synechococcus RF-1
Hso-Freng Yuan*, Chiung-Min Wang, and Han-Wei Kung
Institute of Botany, Academia Sinica, Taipei, Taiwan 11529, Republic of China
(Received March 24, 2000; Accepted July 17, 2000)
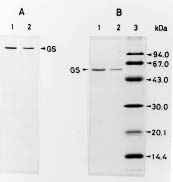
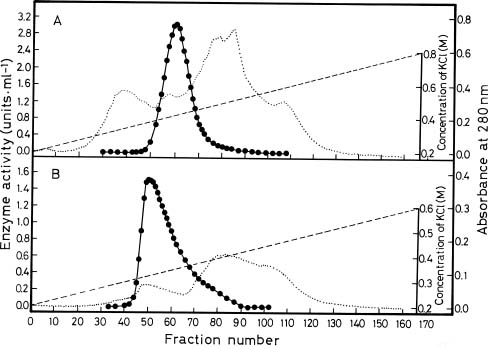
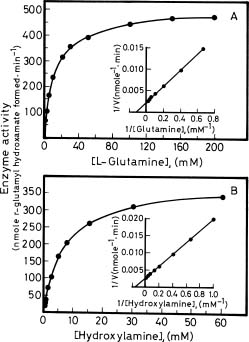
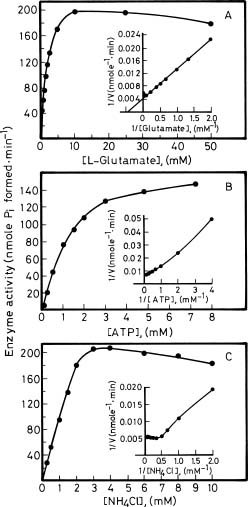
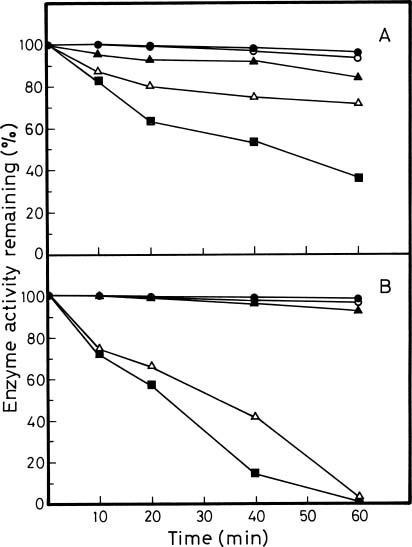
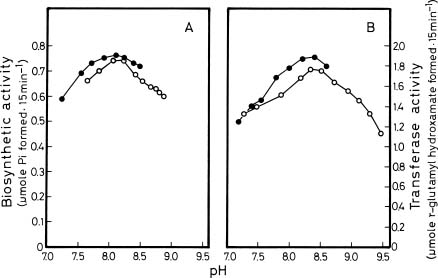
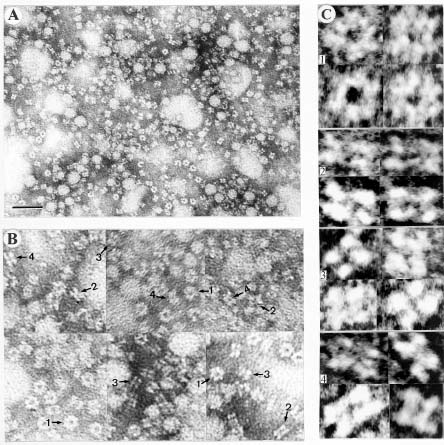
Abstract. Glutamine synthetase (GS; EC 6.3.1.2) from the Synechococcus RF-1 was purified to homogeneity by ion exchange, molecular sieving, and hydroxyapatite chromatographies. The native enzyme has a molecular mass of about 456 kDa, and the molecular mass of its subunit was about 56 kDa. Electron micrographs of the enzyme revealed two parallel protein layers in cubic symmetry with quaternary structure. The actual data indicated that the enzyme could consist of eight identical subunits. The enzyme had an apparent Km value for L-glutamate of 2.33 mM, but it exhibited positive cooperativity for ATP (nH = 1.5 and S0.5 = 0.94 mM) and NH4Cl (nH = 2, and S0.5 = 1.33 mM) in the biosynthetic assay. The enzyme had apparent Km values for L-glutamine and hydroxylamine of 8.70 mM and 7.04 mM, respectively, in the transferase assay. This enzyme was quite stable in Tris-HCl buffer (pH 7.5) containing EDTA, MgCl2 and 2-mercaptoethanol. The pH optima for both the biosynthetic and transferase activities of the enzyme were 8.1 and 8.4, respectively. The enzyme required a divalent metal ion as an activator. Mg2+ was the most effective metal ion for biosynthetic activity, followed by Co2+. Mn2+ was the most effective metal ion for transferase activity. Ca2+ and Mn2+ strongly inhibited Mg2+-supported biosynthetic activity, but Co2+ stimulated it.
Keywords: Cyanobacteria; Glutamine synthetase; Synechococcus RF-1.
Abbreviations: DEAE, diethylaminoethyl; EDTA, ethylenediamine tetraacetic acid; EPPS, N-(2-hydroxymethyl)piperazine-N'-3-propanesulfonic acid; GS, glutamine synthetase; Pi, inorganic phosphate; SDS, sodium dodecyl sulfate; TCA, trichloroacetic acid; Tris, tris(hydroxymethyl)aminomethane.
Introduction
Glutamine synthetase (GS; EC 6.3.1.2) in collaboration with glutamate synthase (EC 1.4.7.1) plays a central role in ammonia assimilation in a wide range of cyanobacteria (Guerrero and Lara, 1987). GS carries out the ATP-dependent synthesis of glutamine from ammonium nitrogen and glutamate while glutamate synthase carries out the reductive transamidation from glutamine to a-ketoglutaric acid that assimilates one molecule of ammonium nitrogen. The incorporation of ammonium nitrogen into carbon skeletons takes place a key junction between nitrogen and the carbon metabolism. Although it does not directly link amino acids with the carbohydrate metabolism, it influences their connection by regulating the glutamine/a-ketoglutarate ratio.
GSs from both N2-fixing and non-N2-fixing cyanobacteria, such as Anabaena (Sampaio et al., 1979; Orr et al., 1981), Anacystis nidulans (Emond et al., 1979; Florencio and Ramos, 1985), Phormidium (Sawa et al., 1988; Blanco et al., 1989) and Synechocystis (Mérida et al.,
1990) have been purified and well characterized. All of these GSs were quite similar in molecular mass, subunit composition, and the requirement of divalent metal ions for enzyme activity. The activity of GSs from Escherichia coli (Shapiro and Stadtman, 1970a) and other gram-negative bacteria (Johansson and Gest, 1977) are regulated by covalent modification through adenylylation/de-adenylylation, but the regulation of cyanobacterial GS activity does not follow the classical adenylylation mechanism present in many prokaryotes. There is no evidence of covalent modification of GS from any cyanobacterium (Fisher et al., 1981). By contrast, the activity of Synechocystis 6803 GS is controlled by a different mechanism that involves the direct interaction of two inhibitory polypeptides with the GS (García-Domínguez et al., 1999).
The unicellular cyanobacterium Synechococcus RF-1 fixes N2 aerobically either under continuous light or in an alternating light/dark cycle (Grobbelaar et al., 1986; Huang and Chow, 1986). Under continuous light, the culture fixes N2 continuously at a variable rate. In a diurnal light/dark regimen, it fixes N2 at a high rate but almost exclusively during the dark periods. In this paper, we describe a complete purification method of GS from the Synechococcus RF-1. We also discuss the properties of the RF-1 GS and the effect of divalent metal ions on enzyme activity.
*Corresponding author. Tel: 886-2-27899590; Fax: 886-2-27827954.